Labs
Explore the Integrative Medical Sciences Department faculty research laboratories.
Faculty Researchers
Professor
Phone: 330.325.6537
Email: ychen1@neomed.edu
Publications
Our research focuses on the molecular mechanisms underlying redox signal pathway in mediating the disease pathogenesis of myocardial infarction. We are interested in the role of mitochondria-derived oxygen free radicals, signals of glutathione and nitric oxide in regulating oxidative post-translational modifications (OPTM) and how these events impact the bioenergetics function of mitochondria in the post-ischemic heart. One line in our work has established alterations of protein S-glutathionylation in mitochondrial complex I and complex II are linked to mitochondrial dysfunction caused by oxidant stress after myocardial ischemia and reperfusion. In collaboration with the principle investigators of NEOMED/KSU, we are extending the research of this marker to the animal models of type II diabetics and obesity.
A second area of work in the lab is focused on the redox pathway of GSH in controlling the status of complex I/complex II S-glutathionylation and superoxide generation mediated by electron transport chain during myocardial ischemia and reperfusion. The mechanism underlying if protein S-glutathionylation as an indicative of overproduction of superoxide in vivo or not is extensively explored. We employ the pharmacologic approach and genetic modified mouse model to study the mechanism of altering redox modification of mitochondrial proteins. We employed the technique of electron paramagnetic resonance (EPR) to measure the oxygen free radical(s) and redox status of mitochondria in heart.
A third area of work in the lab is focus on how eNOS signal pathway regulates mitochondrial function in the heart and the OPTM of mitochondrial electron transport chain in the post-ischemic heart. In collaboration with the investigators from the OSU, we has established that increasing protein tyrosine nitration of complex I and complex II was detected in the post-ischemic heart, and closely related to overproduction of NO by eNOS in the early phase of reperfusion. Physiologically the signal of NO generated by eNOS can regulate mitochondrial respiration, and further modulates the redox status of mitochondria in myocytes. We employ the genetic modified mouse and generate a new mouse model to investigate how the signal by eNOS regulates complex I/complex II S-glutathionylation, and related mitochondrial dysfunction resulted from post-ischemic injury.
Professor
Phone: 330.325.6426
Email: wchilian@neomed.edu
Publications
My interest in the vascular biology has been developing for many years along the lines of acute and chronic adaptations of the coronary circulation to physiological and pathophysiological stresses. With regard to chronic adaptations of the coronary circulation, my laboratory was the first to show that ischemia, rather than shear stress, initiates coronary collateral growth. Shear stress is not precluded from this process; rather this mechanism plays a role in the expansion of the vessel after the ischemic stimulus wanes. More recently my laboratory has studied the role the mitochondrial oxidative stress in coronary collateral development, and we have found that such stress blunts the adaptive growth of coronary collaterals. Moreover, rectification of oxidative stress will restore collateral growth in a preclinical model with a poor growth phenotype. With regard to acute adaptations of the coronary circulation to physiological and pathophysiological stresses, I am proud of the accomplishments of my laboratory: we were the first to document myogenic and flow-dependent control of coronary tone; the distribution of microvascular resistance in the beating heart, the effects of preconditioning extended to the vascular endothelium, the mechanism of alpha-adrenergic coronary constriction involves release of a cardiac myocyte derived vasoconstrictor, coronary metabolic dilation is a feed-forward process involving mitochondrial production of H2O2, metabolic dilation is mediated by redox reactions, and Kv1.5 channels play a critical role in metabolic dilation in the heart.
Associate Professor of Molecular Virology and Cancer Biology
Phone: 330.325.6135
Email: ald@neomed.edu
Publications
My research studies are concerned with the understanding of how certain human papillomaviruses (HPVs) induce uterine cervical cancer, a major cause of women mortality worldwide. There are now known to be a number of other cancers associated with these viruses, such as head and neck tumors and various skin cancers.
Uncontrolled expression of papillomavirus oncogenes may be the spark to ignite the process toward carcinogenesis. Specific topics of my investigation include molecular analysis of how HPV gene expression is regulated by viral and cellular transcription factors. This analysis includes study of a complex regulatory region within the viral genome that contains the major promoter and enhancer elements for the viral oncogenes called the E6 and E7 genes.
Recently, in the process of exploring facets of the viral life cycle, we have discovered that a derivative of the plant lignan, nordihydroguaiaretic acid, has specific and potent inhibitory activity on HPV oncogene expression. Using these compounds, we should be able to reduce the levels of the viral E6 protein and thus interfere with a major route that leads to HPV- induced pre-cancerous and cancerous lesions.
These results, with others, suggest that it may be possible to manipulate the p53 tumor suppressor pathway to induce cell cycle growth arrest and subsequently apoptosis of pre-cancer and cancer cells treated with the plant lignans or in combination with other drugs. In order to determine that we are inducing senescence and/or apoptosis, we are currently measuring telomerase activity within drug-treated cells, cell cycle regulatory proteins, and apoptosis pathway activation.
Associate Professor, Department of Integrative Medical Sciences
Phone: 330.325.6138
Email: fdong@neomed.edu
Publications
My research has focused on exploring the molecular mechanisms underlying cardiovascular diseases, diabetes, and gene and cell therapy for cardiac dysfunction. Additionally, I am responsible for overseeing the Single Cell Genomics facility. Our team examined the role of leptin and adiponectin in obesity-related myocardial dysfunction and established a link between leptin levels and cardiac function. We developed a system with which we can specifically knock down the expression of a gene of interest in White blood cells (WBC)/Hematopoietic stem cells (HSCs). With this system, we found that down-regulation of WBC HIF-1α leads to decreased WBC recruitment and improved left ventricular remodeling following myocardial infarction. In addition, our research reveals a specific paracrine mechanism of Mesenchymal stem cells (MSCs) engraftment. We highlighted the significance of the SDF-1: CXCR4 axis in tissue repair and proposed its exploitation to enhance outcomes in heart failure patients. By developing clinically relevant strategies to manipulate this axis, we can improve outcomes in clinical populations. Moreover, we discovered a new mouse model for aortic valve stenosis, linking CXCR4 deletion in endothelial cells to the development of stenosis and left ventricular hypertrophy. These findings have implications for understanding and potentially treating these cardiac conditions.

Jessica Ferrell, Ph.D.
Phone: 330-325-6468
Email: jfrancl@neomed.edu
www.ferrell-lab.com
The Ferrell Lab investigates the interactions between the liver, intestine, and brain in the development of alcohol-associated liver disease and metabolic syndrome. These organ systems are influenced by the consumption of high fat diets or alcohol, and by circadian disruption like shift work or jet lag, which can worsen disease conditions. Our research goals are to study 1) how alcohol-associated liver disease and metabolic syndrome affect liver function, and 2) how diet and/or circadian disruption contribute to the development of liver disease.
Current work
Project 1
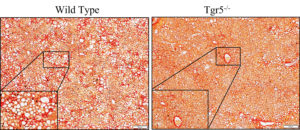
It’s estimated that about 25% of the global population are living with steatosis (fat deposition in the liver), which often occurs along with obesity, Type 2 diabetes, and dyslipidemia. Steatosis, while relatively benign, can progress to steatohepatitis (fatty liver + inflammation), or fibrosis & cirrhosis (scarring) if left untreated. We use special diets to study the transition of steatosis to more severe liver injury states; our goals are to prevent this progression and discover new ways to treat these diseases.
Project 2
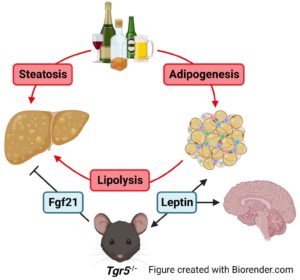
Alcohol consumption can cause steatosis and lead to more severe liver disease like cirrhosis, fibrosis, or alcohol-associated hepatitis, and can also affect the intestine and brain. We are studying the effects of alcohol on liver function as well as the factors that affect alcohol consumption in mice.
Project 3
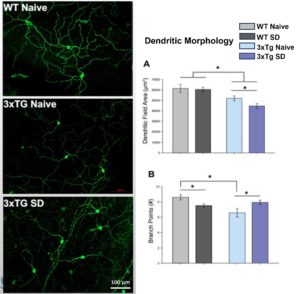
Circadian rhythms direct the timing of our bodies. Disruptions to circadian rhythms (in the form of shift work, jet lag, or sleep loss) can negatively affect organ function and lead to worsened disease states. Currently, we are investigating the effects of long-term sleep deprivation on the progression of conditions like Alzheimer’s disease and metabolic syndrome in mice.
Select publications:
- Pokhrel S, Dilts M, Stahl Z, Boehme S, Frame G, Chiang JYL, Ferrell JM. Tgr5−/− mice are protected from ethanol-induced metabolic alterations through enhanced leptin and Fgf21 signaling. 2023. Commun. 7: e0138. PMID: 37185802.
- Ferrell JM, Dilts M, Stahl Z, Boehme S, Pokhrel S, Wang X, Chiang JYL. Altered serotonin metabolism in Takeda G protein-coupled receptor 5 knockout mice protects against diet-induced hepatic fibrosis. 2022. Res. 6: 214-226.
- Ferrell JM, Chiang JYL. Bile acid receptors and signaling crosstalk in the liver, gut, and brain. 2021. Res. 5: 105-118.
- Chiang JYL, Ferrell JM. Bile acid receptors FXR and TGR5 signaling in fatty liver diseases and therapy. 2020 J. Physiol. Gastrointest. Liver Physiol. 318(3):G554-G573. PMID: 31984784.
- Ferrell JM, Pathak P, Boehme S, Gilliland T, Chiang JYL. Deficiency of both farnesoid X receptor and Takeda G protein-coupled receptor 5 exacerbated liver fibrosis in mice. 2019 Hepatology. 70: 955-970. PMID: 30664797.
- Ferrell JM, Chiang JYL. Understanding bile acid signaling in diabetes: from pathophysiology to therapeutic targets. 2019 Diabetes Metab. J.43(3): 257-272. PMID: 31210034.
- Ferrell JM, Boehme S, Li F, Chiang JY. Cholesterol 7α-hydroxylase-deficient mice are protected from high-fat/high-cholesterol diet-induced metabolic disorders. 2016 Lipid. Res.57: 1144-1154. PMID: 27146480.
- Ferrell JM, Chiang JY. Short-term circadian disruption impairs bile acid and lipid homeostasis in mice. 2015 Cell Mol. Gastroenterol. Hepatol. 1(6): 664-677. PMID: 26645046.
Adam G. Goodwill, Ph.D., F.C.V.S.
Phone: 330.325.6886
Email: agoodwill@neomed.edu
Research in our group focuses on the interplay between myocardial contractile function and delivery of oxygen/nutrients via coronary blood flow. In other words, we study how the heart balances delivery of fuel (blood) with its current demands. Our overall goal is to improve the understanding of key regulators that link oxygen demand with delivery while also exploring how this delicate balance can become disrupted in disease states. Our laboratory employs a top-down approach, beginning at the level of dysfunction and investigating which dysregulated mediators may be responsible for the imbalances associated with cardiac pathologies. Our studies routinely begin in vivo and progress to the molecular. Current work is focused on identifying the mechanisms responsible for cardioprotective phenomena found with certain drug classes. The goal of this work is to identify therapeutic targets for the treatment of cardiovascular disease.
Publications
- Baker HE, Tune JD, Mather KJ, Blaettner BS, Clark HE, Li F, Li X, Kowala MC, Fliegel L, Goodwill AG. Acute SGLT-2i treatment improves cardiac efficiency during myocardial ischemia independent of Na(+)/H(+) exchanger-1. Int J Cardiol. 2022 Sep 15;363:138-148. doi: 10.1016/j.ijcard.2022.06.054. Epub 2022 Jun 23. PubMed PMID: 35753619.
- Goodwill AG, Baker HE, Dick GM, McCallinhart PE, Bailey CA, Brown SM, Man JJ, Tharp DL, Clark HE, Blaettner BS, Jaffe IZ, Bowles DK, Trask AJ, Tune JD, Bender SB. Mineralocorticoid receptor blockade normalizes coronary resistance in obese swine independent of functional alterations in K(v) channels. Basic Res Cardiol. 2021 May 20;116(1):35. doi: 10.1007/s00395-021-00879-3. PubMed PMID: 34018061; PubMed Central PMCID: PMC8552965.
- Tune JD, Baker HE, Berwick Z, Moberly SP, Casalini ED, Noblet JN, Zhen E, Kowala MC, Christe ME, Goodwill AG. Distinct hemodynamic responses to (pyr)apelin-13 in large animal models. Am J Physiol Heart Circ Physiol. 2020 Apr 1;318(4):H747-H755. doi: 10.1152/ajpheart.00365.2019. Epub 2020 Feb 28. PubMed PMID: 32108522.
- Baker HE, Kiel AM, Luebbe ST, Simon BR, Earl CC, Regmi A, Roell WC, Mather KJ, Tune JD, Goodwill AG. Inhibition of sodium-glucose cotransporter-2 preserves cardiac function during regional myocardial ischemia independent of alterations in myocardial substrate utilization. Basic Res Cardiol. 2019 Apr 19;114(3):25. doi: 10.1007/s00395-019-0733-2. PubMed PMID: 31004234; PubMed Central PMCID: PMC6616532.
- Sassoon DJ, Tune JD, Mather KJ, Noblet JN, Eagleson MA, Conteh AM, Sturek JT, Goodwill AG. Glucagon-Like Peptide 1 Receptor Activation Augments Cardiac Output and Improves Cardiac Efficiency in Obese Swine After Myocardial Infarction. Diabetes. 2017 Aug;66(8):2230-2240. doi: 10.2337/db16-1206. Epub 2017 May 8. PubMed PMID: 28483802; PubMed Central PMCID: PMC5521862.
- Goodwill AG, Dick GM, Kiel AM, Tune JD. Regulation of Coronary Blood Flow. Compr Physiol. 2017 Mar 16;7(2):321-382. doi: 10.1002/cphy.c160016. PubMed PMID: 28333376; PubMed Central PMCID: PMC5966026.
- Goodwill AG, Noblet JN, Sassoon D, Fu L, Kassab GS, Schepers L, Herring BP, Rottgen TS, Tune JD, Dick GM. Critical contribution of KV1 channels to the regulation of coronary blood flow. Basic Res Cardiol. 2016 Sep;111(5):56. doi: 10.1007/s00395-016-0575-0. Epub 2016 Aug 5. PubMed PMID: 27496159; PubMed Central PMCID: PMC5193223.
- Goodwill AG, Tune JD, Noblet JN, Conteh AM, Sassoon D, Casalini ED, Mather KJ. Glucagon-like peptide-1 (7-36) but not (9-36) augments cardiac output during myocardial ischemia via a Frank-Starling mechanism. Basic Res Cardiol. 2014;109(5):426. doi: 10.1007/s00395-014-0426-9. Epub 2014 Jul 9. PubMed PMID: 25005062; PubMed Central PMCID: PMC4259250.
Professor of Biochemistry/Molecular Biology
Phone: 330.325.6684
Publications
The cytochrome P450 gene 4 family (CYP4) consists of a group of over 63 members that function to ω-hydroxylate the terminal carbon of fatty acids. In mammals, six subfamilies have been identified and three of these subfamily members show a preference in the metabolism of short (C7-C10)-CYP4B, medium (C10-C16)-CYP4A, and long (C16-C26)-CYP4F, saturated, unsaturated and branched chain fatty acids. These ω-hydroxylated fatty acids are converted to dicarboxylic acids, which are preferentially metabolized by the peroxiosome β-oxidation system to shorter chain fatty acids that are transported to the mitochondria for complete oxidation or used either to supply energy for peripheral tissues during starvation or used in lipid synthesis.
The differential regulation of the CYP4A and CYP4F genes during fasting, by peroxisome proliferators and in non-alcoholic fatty liver disease (NAFLD) and hepatocarcinogenesis suggests different roles in lipid metabolism. The ω-hydroxylation and inactivation of pro-inflammatory eicosanoids by members of the CYP4F subfamily and the association of the CYP4F2 and CYP4F3 genes with inflammatory Celiac disease indicate an important role in the resolution of inflammation. The close clinical association of Celiac disease with inflammatory disease of the liver; primary biliary cirrhosis, primary sclerosing cholangitis, autoimmune hepatitis, hemochromatosis, and fatty liver disease suggest that the CYP4 genes may play a vital functional role in the altered lipid metabolism and inflammation observed in these diseases.
Several human diseases have been genetically linked to the expression CYP4 gene polymorphic variants, which may link human susceptibility to diseases of lipid metabolism and the activation and resolution phases of inflammation. Understanding how the CYP4 genes are regulated during the fasting and feeding cycles and by endogenous lipids will provide therapeutic avenues in the treatment of metabolic disorders of lipid metabolism and inflammation.
My current research is directed toward understanding the control and regulation of the CYP4 genes in fatty liver disease and its progress to steatohepatitis and hepatocarcinogenesis. In fatty liver disease, we hypothesize that induction of the CYP4A genes and uncoupling of the catalytic cycle by excess fatty acids results in increase reactive oxygen species, which is the second hit in the progression of steatosis to steatohepatitis. In contrast, the down-regulation of the CYP4F genes in liver results in both a altered oleic to stearic acid ratio leading to increased fatty acid accumulation in hepatocytes (first hit) and increased pro-inflammatory LTB4 levels leading to neutrophil recruitment to liver (second hit).
Extending this hypothesis to the progression of liver steatohepatitis to hepatocarcinogensis is evident. Down-regulation of CYP4F genes, which metabolize arachidonic acid and pro-inflammatory leukotrienes that recruit immune cells to sites of injury allows transformed cells to escape the host immunosurveillance system and thereby have a selective growth advantage. CYP4F1 gene expression is elevated in hepatic tumors, and cytochrome P4504F1 metabolizes the potent chemokinetic and chemotactic modulator of the inflammatory response, LTB4. Therefore, tumor cells expressing P4504F1 escape the immunological surveillance system.
Patrick T. Kang, Ph.D.
Research Assistant Professor
Office/RGE-331 Lab/RGE-319/320
pkang1@neomed.edu
330-325-6816
Associate Professor
Phone: 330.325.6415
Email: ylee3@neomed.edu
My research interests are the role of orphan nuclear receptor SHP in diet induced diabetes and obesity.

Vahagn Ohanyan, M.D., Ph.D.
Associate Professor
RGE-338 / 330-325-6535
vohanyan@neomed.edu
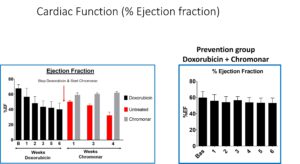
In our laboratory, we are dedicated to investigating the intricate and often poorly understood phenomenon of coronary microvascular dysfunction that occurs in response to chemotherapy. Our primary focus centers on unraveling the mechanisms underlying doxorubicin-induced cardiotoxicity while simultaneously exploring novel treatments and preventive measures to safeguard the heart.
To accomplish these objectives, we have developed a robust preclinical mouse model that mimics the development of heart failure induced by chemotherapy. This model allows us to closely study the pathophysiological changes that occur in the coronary microvasculature and myocardium during chemotherapy administration.
A significant facet of our research involves the utilization of Chromonar, an established medication recognized for its coronary-specific vasodilatory properties. By harnessing the vasodilatory effects of Chromonar, we aim to enhance myocardial blood flow in the presence of doxorubicin, thereby preventing or mitigating the cardiotoxic effects associated with this widely used chemotherapy agent.
Our research endeavors are multifaceted, encompassing detailed molecular and cellular investigations, as well as comprehensive physiological assessments. Through meticulous experimentation and data analysis, we are working to unravel the intricate interplay between chemotherapy, coronary microvascular dysfunction, and the potential protective mechanisms conferred by Chromonar.
The ultimate goal of our research is to not only enhance our understanding of the complex relationship between chemotherapy and cardiac health but also to contribute to the development of more effective therapeutic strategies and prevention methods for chemotherapy-induced cardiotoxicity. By shedding light on these critical issues, we hope to improve the overall quality of life for cancer patients undergoing chemotherapy while ensuring the long-term well-being of their cardiovascular health.
Recent publications:
- Anna Winnicki, James Gadd, Vahagn Ohanyan,Gilbert Hernandez,Yang Wang,Molly Enrick,Hannah McKillen,Matthew Kiedrowski, Dipan Kundu,Karlina Kegecik,Marc Penn,William Chilian,Liya Yin*,Feng Dong* Role of Endothelial CXCR4 in the Development of Aortic Valve Stenosis Original Research, Front. Cardiovasc. Med. – Cardiovascular Therapeutics, DOI: 10.3389/fcvm.2022.971321 Manuscript Id: 971321
- Feng Dong M.D., Ph.D1.,* Liya Yin M.D., Ph.D. 1, Hamayak Sisakian MD., PhD., 2 Tatevik Hakobyan MD., 1 Lacey S. Jeong M.D.1, Hirva Joshi M.D1., Ellianna Hoff M.D1., Selena Chandler M.D1., Geetika Srivastava M.D1., Abdur Rahman Jabir B.S1., Kelly Kimball B.S. 1, Yeong-Renn Chen Ph.D. 1, Chwen-Lih Chen BS1, Patrick T. Kang Ph.D. 1, Parisa Shabani Ph.D. 1, Lindsay Shockling B.S. 1, Thomas Pucci M.S. 1, Karlina Kegecik B.S. 1, Christopher Kolz1, William M. Chilian Ph.D1. and Vahagn Ohanyan M.D., Ph.D1* Takotsubo Syndrome is A Disease of the Coronary Microcirculation- ( European Heart Journal) – (Drs Feng Dong and Vahagn Ohanyan are Co-Corresponding Authors)
- Enrick M, Jamaiyar A, Ohanyan V, Juguilon C, Kolz C, Shi X, Janota D, Wan W, Richardson D, Stevanov K, Hakobyan T, Shockling L, Diaz A, Usip S, Dong F, Zhang P, Chilian WM, Yin L The Roles of Bone Marrow-Derived Stem Cells in Coronary Collateral Growth Induced by Repetitive Ischemia. Cells. 2023 Jan 6;12(2):242. doi: 10.3390/cells12020242.PMID: 3667217
- Khanal S, Bhavnani N, Mathias A, Lallo J, Gupta S, Ohanyan V, Ferrell JM, Raman P. Deletion of Smooth Muscle O-GlcNAc Transferase Prevents Development of Atherosclerosis in Western Diet-Fed Hyperglycemic ApoE-/- Mice In Vivo.Int J Mol Sci. 2023 Apr 26;24(9):7899. doi: 10.3390/ijms24097899.PMID: 37175604
Ravi K. Adapala1#, Venkatesh Katari1#, Anantha K. Kanugula2, Vahagn Ohanyan2, Sailaja Paruchuri1 and Charles K. Thodeti1* Deletion of endothelial TRPV4 protects heart from pressure-overload-induced hypertrophy. Hyperte
Heather A. O’Leary, Ph.D.
Assistant Professor
RGE 244 (Office), RGE 100/113 (Lab)
(330) 325-6868
Associate Professor of Physiology and Pharmacology
Phone: 330.325.6425
Email: praman@neomed.edu
Elucidation of cellular and molecular mechanism(s) of accelerated atherogenesis among diabetics is the primary focus of my research.
In the light of the importance of obesity as an independent risk-factor for both diabetes and related vascular complications, my research program is two-fold: 1) regulation of vascular gene expression by adipokines to better comprehend the role of the adipose tissue in vascular diabetic complications, and 2) to explore the underlying MOA of antidiabetic agents implicated in diabetes and related vascular disorders.
Currently, we are studying the role of thrombospondin-1 (TSP-1), a potent antiangiogenic and proatherogenic protein with strong implications in atherogenesis, in leptin-mediated macrovascular complications.
We are using various approaches that involve cell and molecular biology, biochemistry and mouse models for these studies.
Director, Heart & Blood Vessel Disease Research Area
Associate Professor of Integrative Medical Sciences
Phone: 330.325.6521
Email: lyin@neomed.edu
Current main focus is to study stem cells in cardiovascular regeneration in metabolic syndrome. We study the partially reprogrammed vascular progenitor cells, bone marrow derived stem cells, adipose derived stem cells, cardiac stem cells and induced pluoripotent stem cells in the animal models for coronary collateral growth which will prevent the myocardial ischemia injury or deliver the promising stem cells with biomaterial for tissue regeneration in myocardium infarction injury. We also move forward to clinical translation study in patients with cardiovascular diseases.
Associate Professor of Molecular Pharmacology
Phone: 330.325.6412
Email: jyun@neomed.edu
 Professor of Integrative Medical Sciences
Professor of Integrative Medical Sciences
Adjunct Associate Professor of Pharmaceutical Sciences
Phone: 330.325.6693
Email: yzhang@neomed.edu
Publications
Research in Dr. Zhang’s group aims to understand how bile acid, lipid and glucose metabolism is regulated under physiological and pathological conditions. Dysregulation of bile acid, lipid and/or glucose metabolism may contribute to the pathogenesis of fatty liver disease, diabetes, obesity and atherosclerosis. Dr. Zhang’s group is working on different metabolic targets, including nuclear hormone receptors (FXR, HNF4a, RARa), carboxylesterases, microRNAs, long non-coding RNAs, etc. These targets may play a role in the pathogenesis of metabolic disorders or may serve as therapeutic target(s) for treatment of the common metabolic diseases.
Recent representative publications from Dr. Zhang’s group:
- Xu Y, Hu S, Jadhav K, Zhu Y, Pan X, Cassim Bawa F, Yin L, Zhang Y. Hepatocytic Activating Transcription Factor 3 Protects Against Steatohepatitis Via Hepatocyte Nuclear Factor 4α. Diabetes. 2021 Sep 2;. doi: 10.2337/db21-0181. [Epub ahead of print] PubMed PMID: 34475098.
- Xu Y, Zhu Y, Hu S, Pan X, Bawa FC, Wang HH, Wang DQ, Yin L, Zhang Y. Hepatocyte miR-34a is a key regulator in the development and progression of non-alcoholic fatty liver disease. Mol Metab. 2021 Sep;51:101244. doi: 10.1016/j.molmet.2021.101244. Epub 2021 Apr 28. PubMed PMID: 33930596; PubMed Central PMCID: PMC8141777.
- Xu Y, Zhu Y, Hu S, Xu Y, Stroup D, Pan X, Bawa FC, Chen S, Gopoju R, Yin L, Zhang Y. Hepatocyte Nuclear Factor 4α Prevents the Steatosis-to-NASH Progression by Regulating p53 and Bile Acid Signaling (in mice). Hepatology. 2021 Jun;73(6):2251-2265. doi: 10.1002/hep.31604. Epub 2021 May 14. PubMed PMID: 33098092; PubMed Central PMCID: PMC8062586.
- Xu Y, Li Y, Jadhav K, Pan X, Zhu Y, Hu S, Chen S, Chen L, Tang Y, Wang HH, Yang L, Wang DQ, Yin L, Zhang Y. Hepatocyte ATF3 protects against atherosclerosis by regulating HDL and bile acid metabolism. Nat Metab. 2021 Jan;3(1):59-74. doi: 10.1038/s42255-020-00331-1. Epub 2021 Jan 18. PubMed PMID: 33462514; PubMed Central PMCID: PMC7856821.
- Xu Y, Xu Y, Zhu Y, Sun H, Juguilon C, Li F, Fan D, Yin L, Zhang Y. Macrophage miR-34a Is a Key Regulator of Cholesterol Efflux and Atherosclerosis. Mol Ther. 2020 Jan 8;28(1):202-216. doi: 10.1016/j.ymthe.2019.09.008. Epub 2019 Sep 12. PubMed PMID: 31604677; PubMed Central PMCID: PMC6952168.
- Xu Y, Zalzala M, Xu J, Li Y, Yin L, Zhang Y. A metabolic stress-inducible miR-34a-HNF4α pathway regulates lipid and lipoprotein metabolism. Nat Commun. 2015 Jun 23;6:7466. doi: 10.1038/ncomms8466. PubMed PMID: 26100857; PubMed Central PMCID: PMC4479415.
Contact
Alysia Mulhollen
Business Manager
amulhollen@neomed.edu
330.325.6429
Office: RGE-332
Department Interim Chairman
J. G. M. ‘Hans’ Thewissen, Ph.D.
Phone: 330.325.6295
Email: thewisse@neomed.edu
Department of Integrative Medical Sciences